The preservation of life relies on the flawless synchronization of millions of chemical reactions occurring every second, all sustained within a thermal equilibrium that prevents cell damage. Thermodynamically feasible reactions would take thousands of years to complete in biological timeframes without the presence of enzymes. For example, the hydrolysis of urea takes about 20 years in the absence of a catalyst, whereas the enzyme urease accelerates this reaction by a factor of 10 over 14.
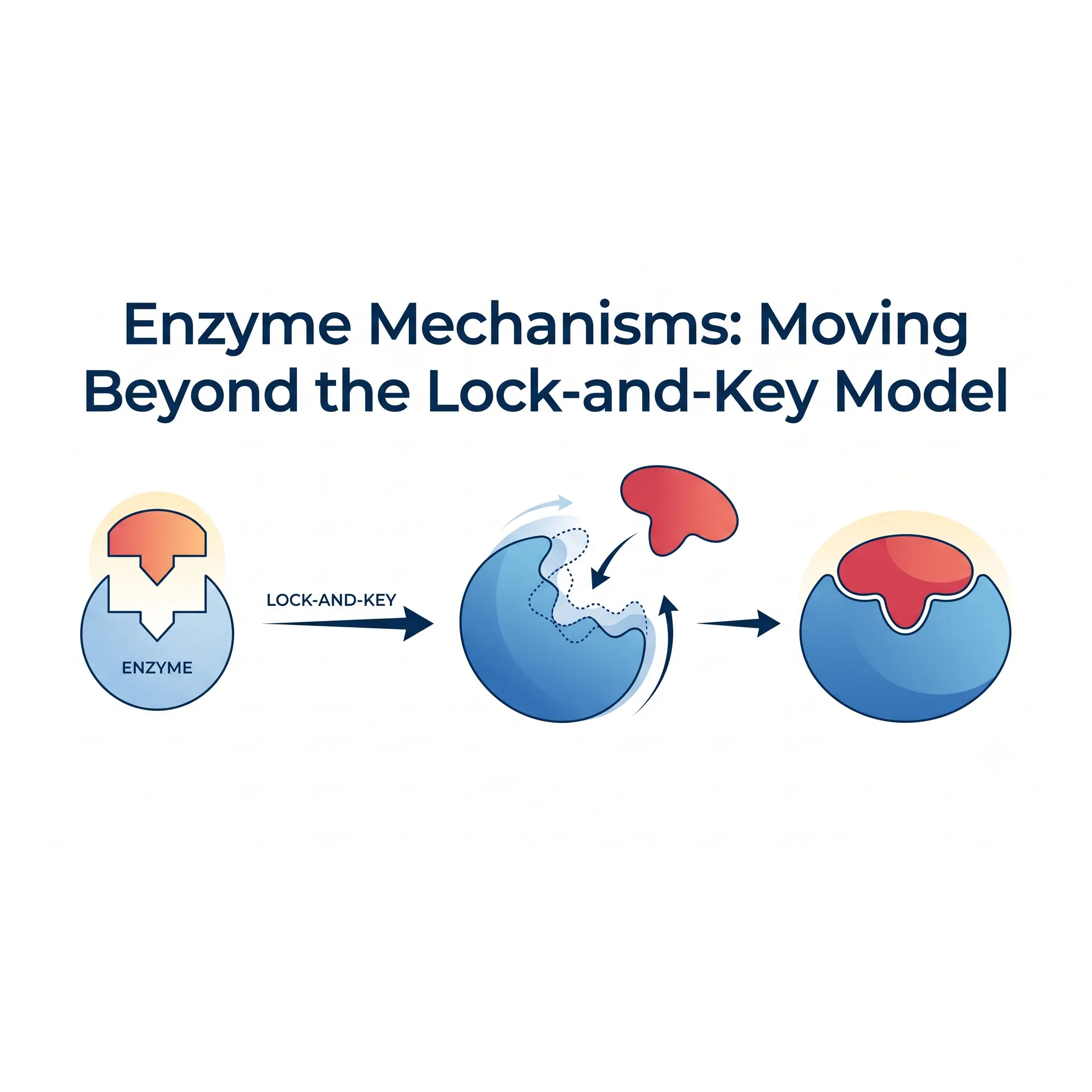
For over a century, the most popular metaphor used in textbooks to explain the enzyme-substrate relationship has been Emil Fischer’s "Lock-and-Key Model." However, modern structural biology, X-ray crystallography, and fast-kinetic studies have revealed that this model is far too primitive to explain the true genius of enzymatic catalysis. Enzymes are not static locks; they are dynamic macromolecular nano-machines that undergo conformational transitions upon binding, twisting and manipulating substrates at the quantum level.
This academic guide explores the true molecular mechanisms behind the catalytic power of enzymes, the induced-fit theory, and the thermodynamic foundations of transition-state stabilization.
Limitations of the Classical Model: Why the Lock-and-Key Theory Fails
Proposed by Emil Fischer in 1894, the Lock-and-Key model asserts that the enzyme's active site possesses a rigid, static geometry that is perfectly complementary to the shape of the substrate. While this model successfully explains enzymatic specificity (selectivity), it falls into a thermodynamic paradox when attempting to account for catalytic velocity and the driving force of the reaction.
If an enzyme were a perfect lock tailored precisely to the substrate's ground state, the enzyme-substrate (ES) complex would fall into a severe thermodynamic energy well, drastically stabilizing the substrate. This stabilization would raise the activation energy barrier required for the reaction to proceed, rather than lowering it. In other words, a flawless lock-and-key fit would stall a chemical reaction rather than accelerate it.
Dynamic Transformations: Koshland and the Induced-Fit Model
In 1958, Daniel Koshland advanced the Induced-Fit Model, proposing that enzymatic specificity and catalysis rely on conformational flexibility rather than static rigidity.
According to this model:
- The active site of the enzyme is not initially a 100% perfect match for the substrate.
- As the substrate approaches the active site, weak non-covalent interactions (hydrogen bonds, van der Waals forces, and electrostatic interactions) trigger a conformational rearrangement in the enzyme's tertiary structure.
- The enzyme reshapes around the substrate, much like a glove conforming to a hand. This dynamic closure induces a desolvation effect, expelling water molecules from the active site to create an isolated, hydrophobic microenvironment optimized exclusively for the reaction.
The Thermodynamic Heart of Catalysis: Transition-State Stabilization
The legendary chemist Linus Pauling summarized the core secret of enzymatic catalysis in a brilliant insight: "Enzymes are complementary to the reaxion's Transition State, not to the substrate itself."
During a chemical reaction, a substrate must reach the transition state—a transient, highly unstable, maximum-energy state where old chemical bonds are partially broken and new ones are partially formed. The activation energy is the net difference between the ground state of the substrate and this transition state.
Enzymes use functional amino acid side chains within their active sites to bind the transition-state molecule with maximum affinity, stabilizing it. By stabilizing the transition state, the peak of the activation energy barrier is significantly lowered.
Enzymes utilize four primary mechanical strategies to reduce this activation energy barrier:
A. Entropy Reduction (Orientation and Proximity)
When two substrates are free in solution, the probability of them colliding in the correct spatial orientation to react is incredibly low. An enzyme binds and aligns the substrates precisely within its active site. This spatial restriction drastically eliminates the translational and rotational entropy barrier of the reaction.
B. Desolvation
In an aqueous solution, substrates are surrounded by a tightly bound hydration shell of water molecules. As the substrate enters the active site, this water shell is stripped away. The removal of water significantly amplifies the strength of electrostatic forces between the enzyme functional groups and the substrate.
C. Distortion (Strain and Distortion)
During the induced-fit transition, the enzyme physically distorts the substrate, mechanically straining the specific chemical bonds targeted for cleavage. This physical strain forces the molecule to adopt a geometry closer to that of the transition state.
D. General Acid-Base and Covalent Catalysis
Specific amino acids within the active site (e.g., Histidine, Aspartate, Glutamate) act as temporary proton donors (acid catalysis) or proton acceptors (base catalysis) to stabilize unstable charges in the transition state. In some mechanisms, functional groups form transient, highly reactive covalent bonds with the substrate, routing the reaction through an alternative path with a lower energy profile.
Modern Biophysical Approaches: Quantum Tunneling and Enzymatic Dynamics
Moving past classical thermodynamics, modern quantum biology shows that certain enzymes (especially dehydrogenases and transferases) smash speed limits via Quantum Tunneling.
During the transfer of very light particles like hydrogen ions (protons), the proton does not physically climb over the activation energy barrier. Instead, owing to wave-particle duality, it "tunnels through" the barrier, appearing instantaneously on the other side. Millimetric protein vibrations (bovine dynamics) within the enzyme active site have evolved to sync perfectly, squeezing the width of the energy barrier to a distance that allows proton tunneling to occur efficiently.
Conclusion
While Emil Fischer's lock-and-key analogy remains a great starting point for understanding enzyme selectivity, it fails to explain the true nature of catalysis. Enzymes are dynamic protein structures shaped by billions of years of evolution. They embrace their substrates, bend molecular bonds to stabilize the transition state, and even manipulate quantum mechanics when necessary.
Grasping this flexible, dynamic architecture is the foundational framework driving modern pharmacology—enabling the rational design of next-generation drugs (such as transition-state analogs that act as powerful enzyme inhibitors)—and fueling industrial biotechnology in the development of engineered artificial enzymes.
References
- Nelson, D. L., & Cox, M. M. (2017). Lehninger principles of biochemistry (7th ed.). W. H. Freeman and Company.
- Koshland, D. E. (1958). Application of a theory of enzyme specificity to protein synthesis. Proceedings of the National Academy of Sciences, 44(2), 98-104. https://doi.org/10.1073/pnas.44.2.98
- Wolfenden, R., & Snider, M. J. (2001). The depth of chemical time and the power of enzymes as catalysts. Accounts of Chemical Research, 34(12), 938-945. https://doi.org/10.1021/ar000058i
- Klinman, J. P., & Kohen, A. (2013). Hydrogen tunneling links protein dynamics to enzyme catalysis. Annu. Rev. Biochem., 82, 471-496. https://doi.org/10.1146/annurev-biochem-051710-133623


No comments yet. You can leave the first one.