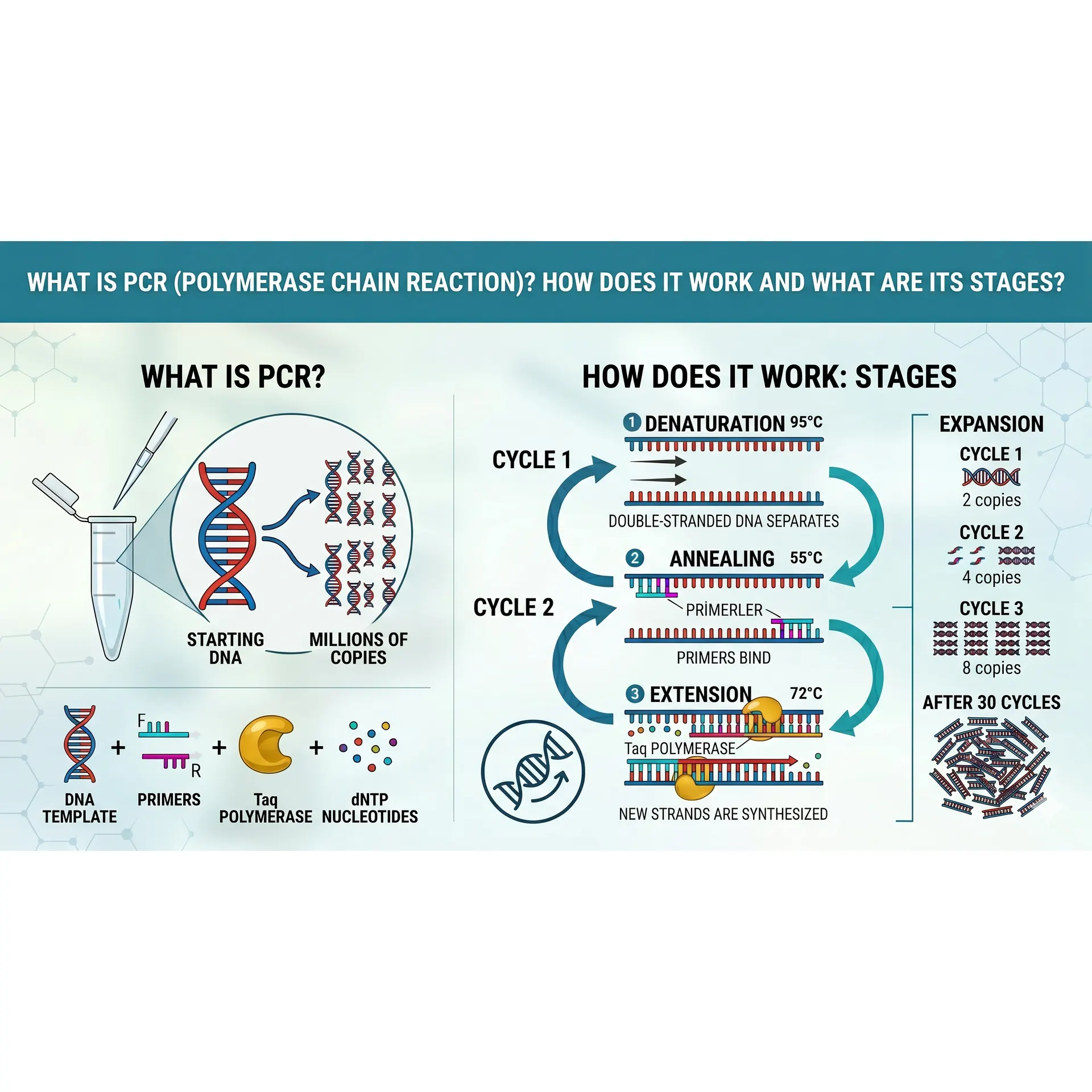
If you are pursuing an undergraduate degree in molecular biology, genetics, bioengineering, or medicine, the most popular acronym you will encounter the moment you step into a laboratory is undoubtedly PCR. Standing for Polymerase Chain Reaction, this technique is universally recognized as the "photocopier" of modern biotechnology.
Developed by Kary Mullis in 1983—a breakthrough that earned him the Nobel Prize in Chemistry in 1993—this method has revolutionized every corner of biology, from forensics and paternity testing to pandemic diagnostics and the evolutionary analysis of extinct organisms.
But how exactly does a tiny, microliter-volume liquid inside a tube replicate a microscopic DNA fragment billions of times over? Let's analyze the mechanics, the chemical cocktail, and the thermal cycling stages of a PCR reaction in full detail through the lens of a molecular biologist.
The Fundamental Logic of PCR
In its simplest definition, PCR is an in vitro (in a tube, outside a living organism) technique used to amplify millions or billions of copies of a specific target DNA sequence using a thermal cycler.
Before a cell divides, it replicates its DNA naturally using complex enzymatic machinery. PCR mimics this natural cellular DNA replication process but does it much faster, with extreme specificity, and entirely in a microcentrifuge tube. At the end of each reaction cycle, the amount of target DNA increases exponentially. Theoretically, if n represents the number of cycles, the number of copies obtained from a single starting double-stranded DNA molecule is calculated using this formula:
copy count = 2 over n
The PCR Master Mix: What’s Inside the Tube?
To execute a successful PCR reaction, specific molecular components must be added into a microcentrifuge tube (commonly prepared as a "PCR Master Mix") in precise, millimetric concentrations:
- Template DNA: The original DNA sample containing the specific target region you want to amplify.
- Taq Polymerase: The high-performance, heat-stable engine of the reaction. Isolated from the thermophilic hot-spring bacterium Thermus aquaticus, this enzyme can withstand extreme temperatures up to 95 C without denaturing (unfolding).
- Primers: Short, chemically synthesized single-stranded DNA sequences (usually 18–25 nucleotides long) that are complementary to the flanks of the target DNA region. A reaction requires a Forward and a Reverse primer. Because DNA polymerase cannot initiate synthesis de novo (from scratch), primers provide the free $3'\text{-OH}$ group required for the enzyme to start building.
- dNTPs (Deoxynucleoside Triphosphates): The raw chemical building blocks used to synthesize the new DNA strand; specifically, a balanced mix of dATP, dCTP, dGTP, and dTTP.
- PCR Buffer and Mg2+ Ions: Provides the optimal pH and ionic strength for maximum enzyme stability. Magnesium chloride (MgCl2) acts as an essential cofactor for Taq polymerase. A deficiency in Mg2+ will stall the reaction, while an excess decreases enzyme fidelity, leading to non-specific binding (amplification of wrong regions).
The 3 Core Stages of a PCR Cycle
A PCR protocol relies on a thermal cycler rapidly switching between three distinct temperatures. These three consecutive steps constitute a single "cycle."
1. Denaturation
- Temperature: 94 - 96 degree
- Duration: 15 - 30 seconds
- Mechanism: The double-stranded template DNA is exposed to high heat, which breaks the weak hydrogen bonds holding the complementary bases together. The double helix unwinds, leaving two separate single strands of DNA. In this in vitro environment, high thermal energy completely replaces the cellular enzyme helicase.
2. Annealing
- Temperature: 50- 65 degree (Strictly calculated based on the melting temperature, or Tm, of the primers).
- Duration: 20 - 40 seconds
- Mechanism: As the temperature drops, the forward and reverse primers, which are present in vast excess in the mix, easily find their complementary sequences on the single-stranded template DNA and form stable hydrogen bonds. If this temperature is set too high, primers cannot bind; if it is too low, primers bind non-specifically to incorrect regions.
3. Extension (Elongation)
- Temperature: 72 degree
- Duration: 30 seconds - 2 minutes (Dependent on the length of the target amplicon; Taq polymerase synthesizes roughly 1,000 base pairs per minute).
- Mechanism: 72 degree is the absolute sweet spot (optimum temperature) for Taq polymerase activity. The enzyme binds to the 3' end of the annealed primers and begins adding free dNTPs from the solution in the 5' -3' direction, synthesizing a brand-new complementary strand.
Once these three stages conclude, one cycle is complete, and the target DNA population doubles. This sequence is typically repeated for 30 to 40 cycles.
Post-PCR Analysis: Visualizing the Product
Once the thermal cycler stops, your tube holds billions of DNA copies, but the liquid remains perfectly clear to the naked eye. To verify if your target amplicon was successfully synthesized, undergraduate labs rely on Agarose Gel Electrophoresis.
Because of the phosphate groups in its backbone, DNA carries a net negative (-) charge. When an electric current is applied across a porous agarose gel, DNA molecules migrate toward the positive (+) anode. Smaller DNA fragments weave through the molecular pores quickly and race ahead, while larger fragments lag behind. When the gel is pre-stained with fluorescent dyes (like Ethidium Bromide or safe alternative dyes) and viewed under a UV transilluminator, the amplified target DNA registers as a sharp, glowing band at a specific base-pair (bp) size mark.
Conclusion
The PCR technique that Kary Mullis visualized while driving along a dark California highway is now the ultimate cornerstone of genetic engineering. Throughout your undergraduate studies, almost every advanced lab application you perform—be it molecular cloning, DNA sequencing, mutation detection, or gene expression profiling—will have this elegant three-stage thermal cycle at its core. Mastering the balance of these reaction components is the single most important academic asset you can develop to troubleshoot any laboratory hurdle.
References
- Mullis, K., Faloona, F., Scharf, S., Saiki, R., Horn, G., & Erlich, H. (1986). Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harbor Symposia on Quantitative Biology, 51, 263-273. https://doi.org/10.1101/sqb.1986.051.01.032
- Saiki, R. K., Gelfand, D. H., Stoffel, S., Scharf, S. J., Higuchi, R., Horn, G. T., ... & Erlich, H. A. (1988). Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science, 239(4839), 487-491. https://doi.org/10.1126/science.2448875
- Sninsky, J. J., Innis, M. A., & Gelfand, D. H. (Eds.). (2012). PCR protocols: a guide to methods and applications. Academic Press.



No comments yet. You can leave the first one.